SINDROME MARFAN
El síndrome de Marfan es un trastorno hereditario que afecta el tejido conjuntivo, es decir, las fibras que sostienen y sujetan los órganos y otras estructuras del cuerpo. El síndrome de Marfan afecta más frecuentemente el corazón, los ojos, los vasos sanguíneos y el esqueleto.
Las personas con síndrome de Marfan generalmente son altas y delgadas, y tienen brazos, piernas y dedos de pies y manos desproporcionadamente largos. El daño causado por el síndrome de Marfan puede ser leve o intenso.

CAUSAS
El síndrome de Marfan es provocado por un defecto en el gen que le permite al cuerpo producir una proteína que ayuda a darle elasticidad y fuerza al tejido conjuntivo.La mayoría de las personas con síndrome de Marfan heredan el gen anormal del padre que tenga este trastorno. Cada hijo de un progenitor afectado tiene una probabilidad de 50/50 de heredar el gen defectuoso. En alrededor del 25 por ciento de las personas con síndrome de Marfan, el gen anormal no proviene de ninguno de sus padres. En estos casos, se desarrolla una nueva mutación de forma espontánea.
Complicaciones cardiovasculares
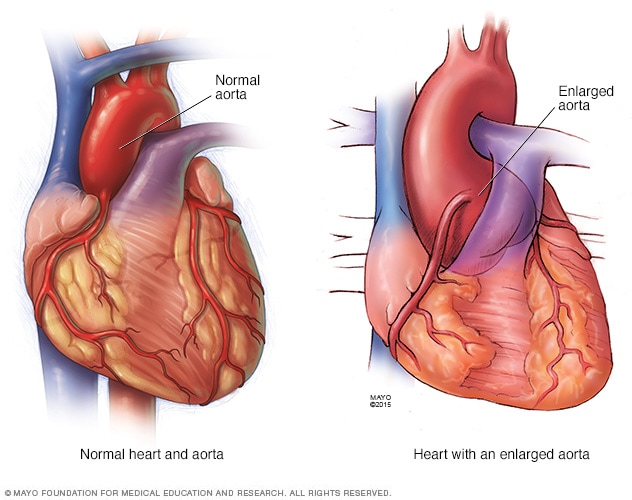
Las complicaciones más peligrosas del síndrome de Marfan afectan el corazón y los vasos sanguíneos. El tejido conjuntivo defectuoso puede debilitar la aorta, la arteria grande que surge desde el corazón y suministra la sangre al organismo.
Complicaciones en los ojos
Las complicaciones en los ojos pueden comprender las siguientes:
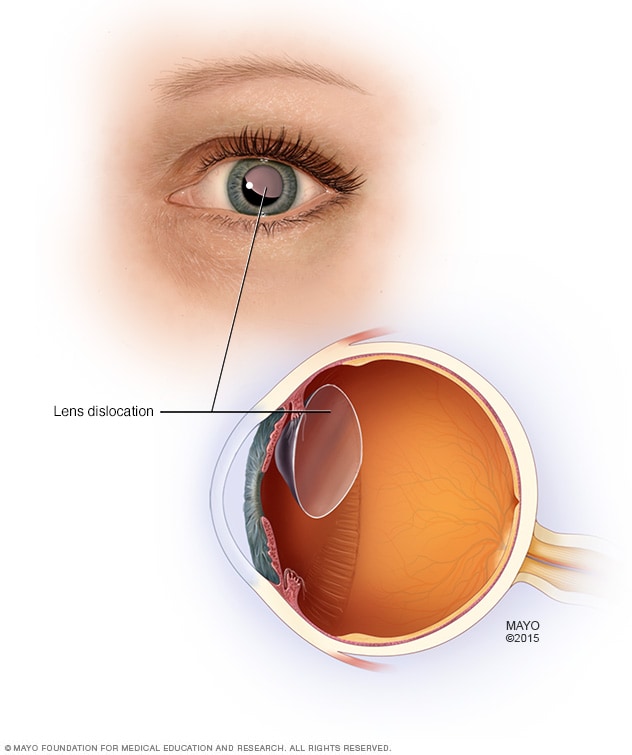
- Luxación del cristalino. La lente de enfoque dentro del ojo puede moverse del lugar si se debilitan sus estructuras de soporte. El término médico para este problema es «ectopia del cristalino», y ocurre en más de la mitad de las personas que tienen síndrome de Marfan
FENOTIPO
M: La afectación mitral consiste en la aparición de prolapso valvular, con diversos grados de regurgitación.
A: Los diámetros de la raíz aórtica pueden estar en los límites superiores de la normalidad para el tamaño del cuerpo, pero no hay progresión a aneurisma ni predisposición a la disección.
S: Aparecen estrías en la piel como las que existen en obesos.
S: Existen anomalías esqueléticas como las del Marfan.

GENOTIPO
50% ENFERMOS
50% SANOS
GEN DONDE SE UBICA:
Se asocia el gen FBN1 del cromosoma 15.El
FBN1 codifica una proteina llamada Fibrilina
que es escencial para la formación de fibras elasticas del tejido conectivo.
Una mutacion en el gen FBN1 puede reducir la cantidad de funciones de la proteina fibrilina.

Tratamiento
En el pasado, la gente que tenía el síndrome de Marfan no solía vivir más de 40 años. Con controles periódicos y un tratamiento moderno, la mayoría de las personas con síndrome de Marfan ahora puede esperar llevar una vida más normal.
En el pasado, la gente que tenía el síndrome de Marfan no solía vivir más de 40 años. Con controles periódicos y un tratamiento moderno, la mayoría de las personas con síndrome de Marfan ahora puede esperar llevar una vida más normal.
Terapia
El cristalino dislocado dentro del ojo puede tratarse de forma efectiva con anteojos o lentes de contacto que refracten alrededor del cristalino o a través de él.
Cirugías y otros procedimientos
Según los signos y síntomas, los procedimientos pueden comprender:
Reparación aórtica. Si el diámetro de la aorta se dilata rápidamente o alcanza aproximadamente las 2 pulgadas (5 cm), el médico puede recomendar realizar una operación para reemplazar una porción de la aorta por un tubo de material sintético. Esto puede ayudar a prevenir una rotura potencialmente mortal. Quizá también sea necesario reemplazar la válvula aórtica.
ESTADISTICAS
Descrito en 1986 por el pediatra francés Antoine Bernard-Jean Marfan, el síndrome de Marfan es una enfermedad hereditaria del tejido conectivo que afecta principalmente a los sistemas cardiovascular, ocular y músculo-esquelético.
La prevalencia estimada es de 1 por cada 5.000-10.000 nacidos vivos, con afectación similar por sexo.
Se hereda con carácter autosómico dominante con alta penetrancia y una marcada heterogeneidad fenotípica (dentro y entre familias afectadas) aunque un 25% de los casos no presenta historia familiar, por lo que se debe a una mutación de novo.
por: Gabriela Cabrera



No hay comentarios.:
Publicar un comentario